A brief introduction to xHMMER3x2
Background & Motivation
Over the past decade, the OMICs frenzy from arrays to sequencing has swarmed genomic research
with voluminous amount of data and elucidated lists of candidate genes/proteins.
Yet, many of these genes/proteins remained not well-understood in relation to the observed phenotype.
The major gap stems from our lack in complete gene/protein functions which impedes researchers
from assembling the sets of biomolecular mechanisms that can sufficiently explain
the observed phenotype.
Despite so, experimental characterization of gene/protein function still receives insufficient attention nowadays.
This can be attested by the dwindling number of characterized genes/proteins reported over the past decade.
Sadly, one can be expected this number to continue to grow at a slow rate.
On this basis, the only viable approach to characterize genes/proteins is to computationally transfer function annotation of the
well-studied gene/protein sequences to the less-studied or novel ones for functional hints.
Besides this, in-silico function annotations must always be kept consistent
regardless of the revisions made to function annotation databases or associated software tools used during the computations.
A widely popular method of functional annotations of proteins is through the domain finding using HMMER software,
with HMMER3 being the latest release. While the local-mode HMMER3 is notable for its massive speed improvement,
the slower glocal-mode HMMER2 is more exact for domain annotation by enforcing full sequence-to-domain alignments.
Since a unit of domain necessarily implies a unit of function, local-mode HMMER3 alone
remains insufficient for precise function annotation tasks.
In addition, the incomparable E-values ranges of the same domain model by different HMMER builds (ie HMMER2 & HMMER3)
creates difficulty when checking for domain annotation consistency on a large-scale basis.
Thus the aim of xHMMER3x2 is to utilize HMMER3's speed and HMMER2's glocal alignment mode for large-scale protein domain annotation.
To put it in simple term, we want to have a "fast" glocal-mode HMMER2.
Methods
The "fast" glocal-mode HMMER2 is achived through a three-stages xHMMER3x2 workflow as shown in Figure 1 below.
Figure 1. The xHMMER3x2 workflow (click on figure itself for larger version)
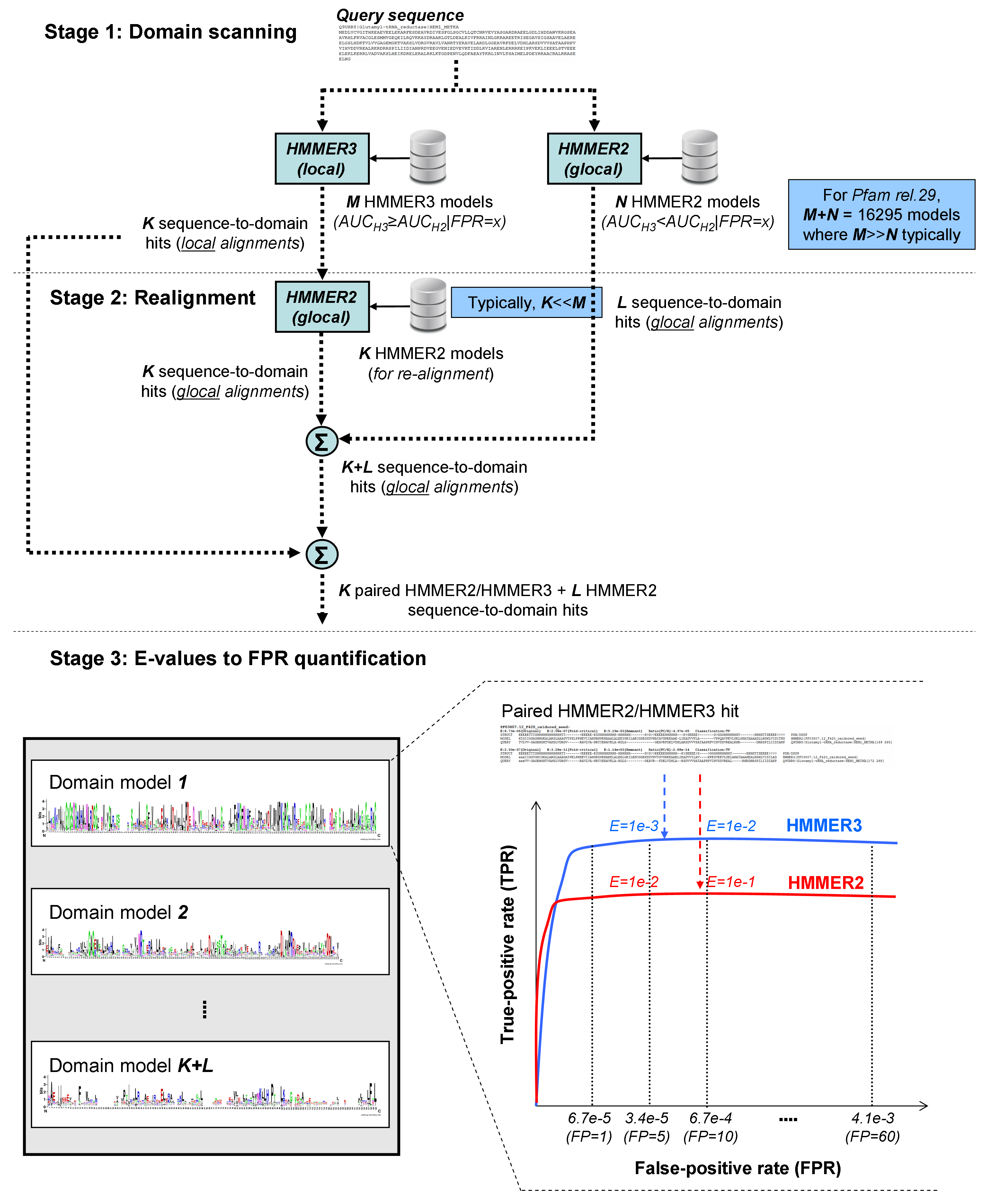
Stage 1: Domain scanning with the bulk of domains scanned by HMMER3
The first stage of xHMMER3x2 is to scan for most probably domains using both HMMER2 & HMMER3. Pfam domains will first be partitioned into 2
portions: one portion of it will be HMMER3 built models while another portion of it will be HMMER2 built models. Using the recommended settings
based on our findings, about 90% of domains (pfam 29) will be assigned for HMMER3 built models, and to be searched by HMMER3,
while the rest 10% of the domains will be assigned for HMMER2 built models, to be searched by HMMER2. As for how the domains are assigned,
they are through the AUC comparison of calibrated HMMER2/HMMER3 ROCs (Figure 2).
 Since the total number of utilized HMMER3 models far exceeds that of HMMER2 (90% versus 10%),
the speed of xHMMER3x2 is expected to be closer to the faster HMMER3 than the slower HMMER2.
Since the total number of utilized HMMER3 models far exceeds that of HMMER2 (90% versus 10%),
the speed of xHMMER3x2 is expected to be closer to the faster HMMER3 than the slower HMMER2.
Detection sensitivity is maintained since a portion of domains
(found to have low sensitivity with HMMER3) is searched by HMMER2 instead.
Stage 2: Full domain re-alignment step with HMMER2
In second stage, domains scanned by HMMER3 is of local alignmnet only. These domains will thus be searched against the same
domain model but in glocal-mode HMMER2 to perform the full domain re-alignment step.
Glocal alignment is prefered in function annotation tasks.
Stage 3: E-values to FPR quantification
The third stage of xHMMER3x2 estimates the FPR(FP) values of the domain hits from their E-values using the
previously calibrated HMMER2/HMMER3 ROCs (Figure 2).
This is used to overcome the problem of incomparable E-values ranges of the same domain model by different
HMMER builds (HMMER2,HMMER3).
The FPR intervals are bounded by these FPR(FP) points:
- 6.74e-5 (1)
- 3.37e-4 (5)
- 6.74e-4 (10)
- 1.01e-3 (15)
- 1.35e-3 (20)
- 1.69e-3 (25)
- 2.02e-3 (30)
- 2.69e-3 (40)
- 3.37e-3 (50)
- 4.05e-3 (60)
- 4.30e-3 (70)
- 4.90e-3 (80)
- 5.50e-3 (90)
- 6.14e-3 (100)
Figure 2. Calibration procedure between HMMER2 and HMMER3 domain model builds (click on figure itself for larger version)
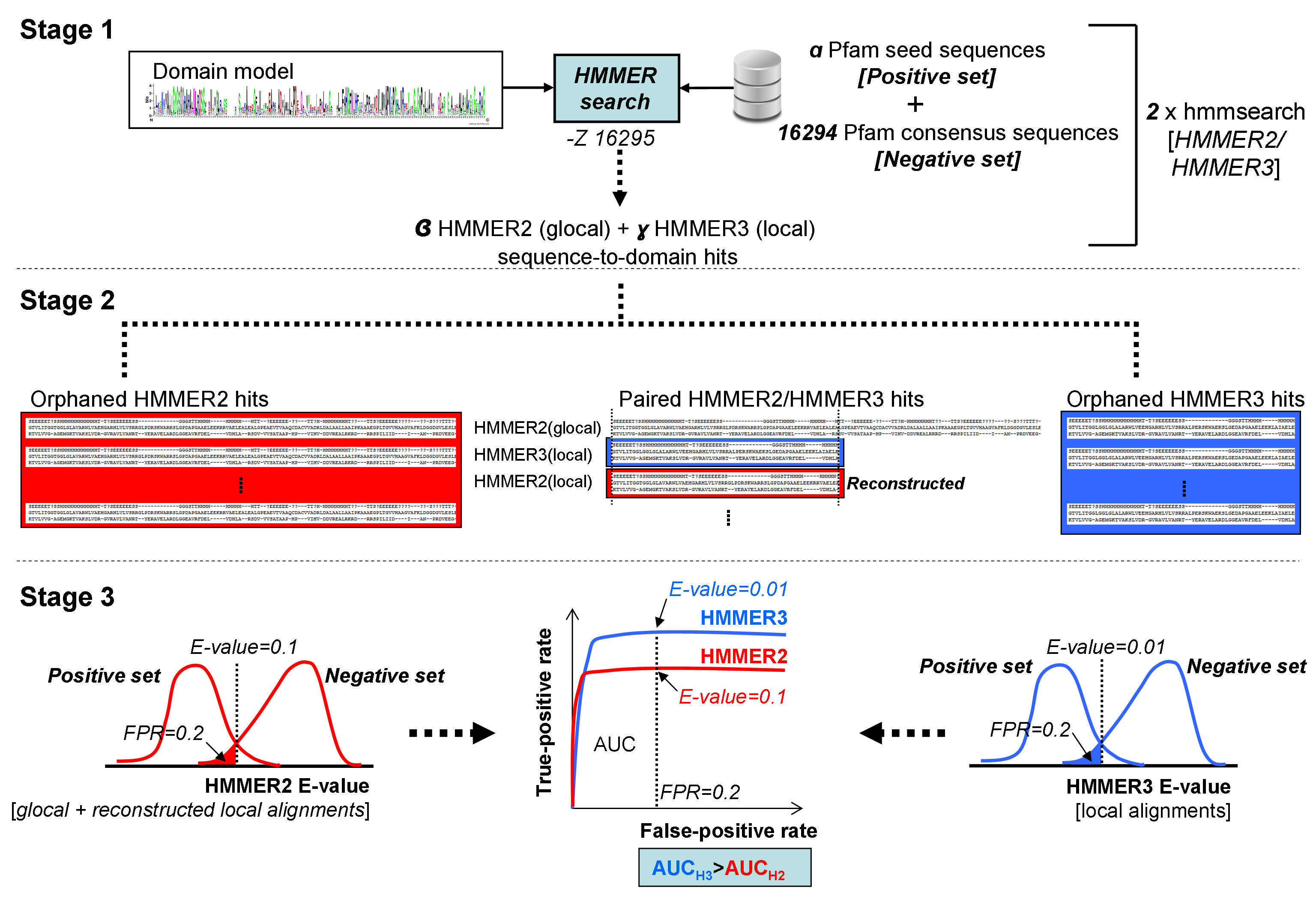
Stage 1
The "hmmsearch" results for the two HMMER variants are first produced on a positive set of α seed sequences
of the model with the rest of the 16294 models (pfam 29) as consensus sequences.
Stage 2
A total of β glocal HMMER2 and γ local HMMER3 sequence-to-domain alignments are paired HMMER2/HMMER3 hits or orphaned accordingly.
For the paired hits, the HMMER2 hit is reconstructed to mimic the local alignment of its HMMER3 counterpart to derived a new HMMER2 E-value.
Stage 3
The orphaned and reconstructed E-values of the HMMER2 are partitioned into their positive and negative sets (see red histograms).
The same is done for the HMMER3 (see blue histograms). For a particular E-value cutoff,
the respective false-positive rates (FPR) and true-positive (TPR) rates can be quantified from the two sets of histograms.
Calibration of HMMER2 & HMMER3 is important in xHMMER3x2 for the following purposes:
- To identified which domains to be of HMMER2/HMMER3 built models (stage 1 of xHMMER3x2). This is done through the AUC comparison of HMMER2 and HMMER3 ROC.
If AUCH3>AUCH2, this particular domain will have a better search result with HMMER3, and vice versa.
- To serve as common metric between HMMER2 and HMMER3 output, eliminating incomparable E-values between the HMMER builds (stage 3 of xHMMER3x2).
Summary
In this work, the speed of HMMER3 and glocal-mode alignment of HMMER2 are combined to create the
xHMMER3x2 framework (Figure 1) to tackle the large-scale domain annotation task.
Briefly, HMMER3 is utilized for initial domain detection so that HMMER2 can subsequently
perform the glocal-mode sequence-to-domain alignments for the detected HMMER3 hits.
A calibration procedure is required to ensure that the search space by HMMER2 is sufficiently
replicated by HMMER3. Generally, the latter is true for 80% of the Pfam domain library (release 29).
However, about 20% of HMMER3 domain models has to be replaced by HMMER2 counterparts if the search space
of HMMER2 is to be closely replicated.
When tested on a set of UniProt/SwissProt human sequences,
xHMMER3x2 can be configured to be between 7 to 201 times faster than HMMER2,
but with descending domain detection sensitivity from 99.8% to 95.7% with respect to HMMER2.
At extremes, xHMMER3x2 is either the slow glocal-mode HMMER2 or fast HMMER3 with glocal-mode.
At recommended setting, xHMMER3x2 can achieve 99.1% sensitivity with 18x speed up.
Finally, the E-values to false-positive rates quantification by xHMMER3x2 allows E-values
of different model builds to be compared, so that any annotation discrepancies in a
large-scale annotation exercise can be flagged for manual examination.
Overall, the xHMMER3x2 workflow allows large-scale domain annotation speed to be improved over HMMER2
without compromising for domain-detection sensitivity and sequence-to-domain alignment incompleteness.